
Principal RESEARCH lines
At the moment, the main fields under investigation are:
Photochemical organic synthesis
- Photocatalyzed reactions
- Reactions of photogenerated aryl cations
- Scale-up of photochemical syntheses
Applied photochemistry
- Preparation of potential chemotherapeutics activated by light
- Aromatic Sulfonates and Phosphates as PhotoAcid Generators (PAGs)
- Photodegradation of biologically active organic compounds
Photochemical organic synthesis
C-C bond formation reactions are one of the main targets in organic synthesis. Since many years, our laboratory has been active in this field, studying the photochemical generation of highly reactive intermediates (such as carbon centered radicals and aryl cations) under mild conditions with no need to use metal catalysts or organometallic species, both expensive and toxic.
Photocatalyzed reactions
Photocatalyzed reactions are a particular class of photochemical processes that occur only in the presence of a catalyst and upon irradiation with light. Photocatalysis was first introduced as a method for the production of hydrogen from water (water splitting) or for the photodepollution of water or air. Nevertheless, in recent years this approach has also been used for the synthesis of organic molecules. [1-3]
Commonly used photocatalysts are:
- Solid semiconductor particles (e.g. TiO2);
- Organic molecules (e.g. benzophenone, cianoaromatics, dyes);
- Inorganic salts (e.g. decatungstate anion).
The main features of photocatalysis are:
- very mild reaction conditions. The use of the (highly reactive) excited state of the photocatalyst avoids the use of any aggressive auxiliary material or the use of high temperatures; this notwithstanding, the method generates radicals from unusual, non activated precursors, such as alkanes and amides;
- Contrary to the usual photochemical approach, which requires the use of light absorbing reagents, the photocatalytic method can be used to activate molecules virtually photochemically unreactive (e.g. alkanes), since the photocatalyst is the only absorbing light species in the reaction mixture.
It is important to stress that all the features previously reported well fit with the main principles of green chemistry. Photocatalysis is thus a really environment-friendly candidate for a valid alternative to the known thermal synthetic strategies.
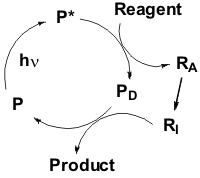
The term photocatalysis comes from the union of the words "photochemistry" and "catalysis". On one hand, the photocatalyst P absorbs the radiation and is chemically active in the excited state, on the other hand it is a part of a catalytic cycle and can be used in a sub-stoichiometric amount, as it happens in thermal catalysis. As for the above, the photocatalytic method (see the Scheme on the left) is based on the use of the photoactive compound P.
This is able to absorb light and, once reached the excited state (P*), it is able to activate (usually by hydrogen abstraction or electron transfer) the reagent (R). The resulting intermediate (RA) then evolves to RI and, after reaction with the deactivated photocatalyst (PD), gives the end product with the concomitant regeneration of P.
Recently, our research group has focused its attention on TBADT (tetrabutylammonium decatungstate). This compound (a white powder) is able to activate the C-H bonds in various molecules by homolytic cleavage of this bond (see the Scheme below) and allows for the generation of radicals (R•) under very mild conditions. The latter intermediate is then trapped by electron-poor olefins (α,β-unsaturated esters, ketones or nitriles) resulting in an overall radical conjugate addition onto a C=C double bond.
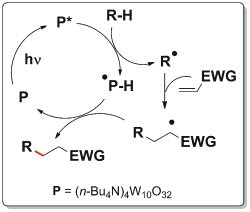
TBADT has proven to be a robust and versatile photocatalyst: it can be used in a small amount (2% eqv.) and remains active for many catalytic cycles. Moreover, it is able to activate many different classes of substrates, otherwise non-reactive. Many compounds have been used as substrates for this reaction: alkanes, alcohols, ethers, acetals, aldehydes and amides. [4]
Future developments
Recently, our attention has been focused on the axploitation of the decatungstate photocatalyst for photoinduced electron transfer reactions (PET, in alternative to the above mentioned hydrogen atom transfer - HAT- mechanism). [5] Another field under investigation concerns with the discovery of innovative photocatalysts able to work under visible light irradiation.
References
- Fagnoni, M.; Dondi, D.; Ravelli, D.; Albini, A. "Photocatalysis for the Formation of the C-C Bond" Chem. Rev. 2007, 107, 2725-2756. DOI: 10.1021/cr068352x
- Ravelli, D.; Dondi, D.; Fagnoni, M.; Albini, A. "Photocatalysis. A multi-faceted concept for green chemistry" Chem. Soc. Rev. 2009, 38, 1999-2011. DOI: 10.1039/B714786B
- Ravelli, D.; Fagnoni, M.; Albini, A. "Photoorganocatalysis. What for?" Chem. Soc. Rev. 2013, 42, 97-113. DOI: 10.1039/C2CS35250H
- Ravelli, D.; Montanaro, S.; Zema, M.; Fagnoni, M.; Albini, A. "A Tin-Free, Radical Photocatalyzed Addition to Vinyl Sulfones" Adv. Synth. Catal. 2011, 353, 3295-3300 and references therein. DOI: 10.1002/adsc.201100591
- Montanaro, S.; Ravelli, D.; Merli, D.; Fagnoni, M.; Albini, A. "Decatungstate As Photoredox Catalyst: Benzylation of Electron-Poor Olefins" Org. Lett. 2012, 14, 4218-4221. DOI: 10.1021/ol301900p
Reactions of photogenerated aryl cations
Aryl cations are electrophilic species can not be thermally generated in solution, but they are smoothly accessible under mild conditions through the photoheterolysis of electron-rich aryl halides (or esters, see Scheme below). [1]
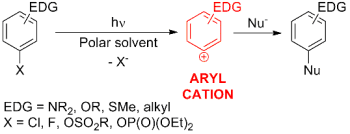
The high reactivity of aryl cations towards different kinds of nucleophiles has been exploited by our research group for the metal-free photochemical formation of aryl-carbon bonds. [2] The Scheme below shows some significant examples of this synthetic strategy.
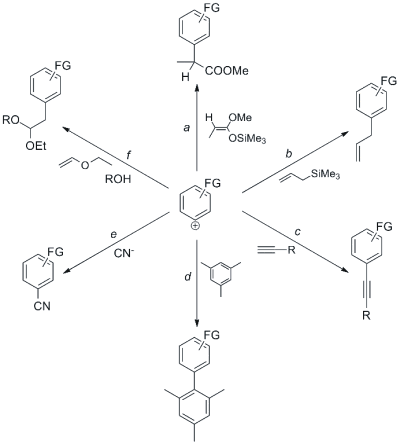
If ketene silyl acetals are used as nucleophiles (path a), the photochemical reaction leads efficiently to the corresponding α-aryl propionic acids. These are interesting molecules, since some compounds bearing this moiety are known as NSAIDs (NonSteroidal Anti-Inflammatory Drugs).
A variety of allyl anisoles and allyl phenols (naturally occurring bioactive compounds) has been obtained in nearly quantitative yields, by reaction of aryl cations with allyltrimethylsilane (path b).
A surprisingly clean reaction occurred upon reactions of substituted phenyl cations with terminal alkynes, where the corresponding aryl alkynes were isolated in 70-90% yield (path c). This process is actually an interesting metal-free alternative to the dimetallic-mediated (palladium and copper) Sonogashira reaction.
We likewise applied phenyl cation chemistry to the arylation of aromatics, such as benzene and symmetric methylbenzenes (path d). Thus, we obtained several asymmetric or crowded biaryls in high yields. Noteworthy, steric hindering caused no major limitation and the arylation was fully chemoselective.
π-Bond nucleophiles are not the only useful traps for aryl cations. For example, such electrophiles are highly reactive also towards inorganic anions, such as iodide and cyanide. The irradiation of aryl halides and esters in a water-acetonitrile solution of KCN afforded various benzonitriles, an important class of compounds which finds application among agrochemicals and pharmaceuticals (path e).
Recently, we have also proposed a new three-component reaction for the synthesis of aryl acetals and aryl ketals; this exploits the reaction of aryl cations with a vinyl ether followed by the addition of the alcoholic medium (path f).
Future developments
Our interest in phenyl cations chemistry is now focusing on the use of different precursors, including a variety of aromatic sulfonates. Moreover, we are pursuing the research for innovative synthetic procedures exploiting these highly reactive intermediates. [3,4]
References
- Fagnoni, M.; Albini, A. "Arylation Reactions: The Photo-SN1 Path via Phenyl Cation as an Alternative to Metal Catalysis" Acc. Chem. Res. 2005, 38, 713-721. DOI: 10.1021/ar0402356
- Dichiarante, V.; Fagnoni, M. "Aryl Cation Chemistry as an Emerging Versatile Tool for Metal-Free Arylations" Synlett 2008, 787-800. DOI: 10.1055/s-2008-1042933
- Raviola, C.; Protti, S.; Ravelli, D.; Mella, M.; Albini, A.; Fagnoni, M. "Acetalization Allows the Photoheterolysis of the Ar–Cl Bond in Chlorobenzaldehydes and Chloroacetophenones" J. Org. Chem. 2012, 77, 9094–9101. DOI: 10.1021/jo3016264
- Protti, S.; Fagnoni, M.; Albini, A. "A Photochemical Route to 2-Substituted Benzo[b]furans" J. Org. Chem. 2012, 77, 6473-6479. DOI: 10.1021/jo3010183
Scale-up of photochemical syntheses
The use of photochemical processes for industrial application is very limited and these reactions hardly express their full synthetic potential. In the aim of developing processes suitable for large scale syntheses, our laboratory is revising and optimizing a number of reaction protocols in view to ameliorate their environmental performance.
Thus, we are focusing our attention on the use of flow conditions and on the replacement of artificial light sources (multi-lamp apparatus or immersion well) with direct sunlight or with a suitable simulator. In both cases, preliminary experiments allowed us to devise simple gram scale syntheses of valuable compounds.
Future developments
Recently, we became interested in the use of low power consumption light sources (such as LEDs) and in the design of photochemical microreactors.
References
- Protti, S.; Ravelli, D.; Fagnoni, M.; Albini, A. "Solar light-driven photocatalyzed alkylations. Chemistry on the window ledge" Chem. Commun. 2009, 7351-7353. DOI: 10.1039/B917732A
- Dondi, D.; Protti, S.; Albini, A.; Mañas Carpio, S.; Fagnoni, M. "Synthesis of γ-lactols, γ-lactones and 1,4-monoprotected succinaldehydes under moderately concentrated sunlight" Green Chem. 2009, 11, 1653-1659. DOI: 10.1039/B904427B
Applied photochemistry
Photochemical processes find many applications other than organic synthesis. These cover diverse fields, from technology to biology. As detailed below, our research group is also involved in some of these.
Preparation of potential chemotherapeutics activated by light
Chemotherapeutic agents must be sufficiently aggressive to kill tumor cells, yet inactive against healthy cells. Among the few chemical classes that come close to these idealized agents are highly unsaturated hydrocarbons such as enyne-allenes. These 10 π electron polyunsaturated systems undergo easy (often spontaneous) cycloaromatization by converting two σ bonds into a π bond (the so-called Myers-Saito cyclization) to form a α,3-didehydrotoluene biradical (α,3-DHT, a σ,π-biradical). These radicals are known to be aggressive and able to abstract hydrogen atoms from deoxyribose residues in DNA, finally causing oxidative DNA cleavage and cell death.
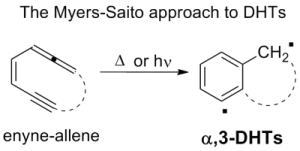
This approach, however, requires a multistep synthesis of complex structures, which limits the overall viability. Furthermore the generation of DHTs from the Myers-Saito cyclization led exclusively to α,3-didehydrotoluenes. We recently developed a completely different approach that obtains the same DHT active intermediates by supplying a large amount of energy to a stable, easily synthesized compound, rather than from an unstable precursor obtained through an expensive synthesis. The breakthrough of our research is arriving to α,n-DHTs by two consecutive eliminations from a ring-substituted benzyl derivative (Ar-LG) via a phenyl cation intermediate. In the enyne-allene system, aromaticity is arrived at the same time as biradical formation, whereas our approach starts from an aromatic, and the required high energy activation is conveniently supplied by irradiation. The adoption of stable precursors broadens the field of accessible structures that can thus be tailor-made for specific actions and, furthermore, the photochemical activation has practical advantages in the application of the therapy.
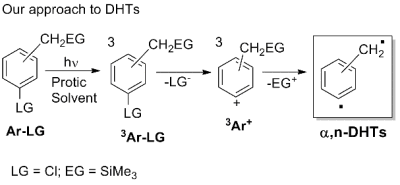
Thus, Ar-LG is an aromatic compound having a leaving group (LG) and a -CH2EG group (EG = an electrofugal group) that reacts under irradiation in solution. In the first step, a triplet phenyl cation (3Ar+) is generated by fragmentation of the triplet excited state of the aromatic (3Ar-LG) and in turn loses the EG group forming the desired (α,n-DHT).
Future developments
We are now planning to synthesize different DHT precursors, characterized by different LG and EG groups. Likewise we are exploring the effect of further functional groups conjugated with the aromatic core.
References
- Protti, S.; Ravelli, D.; Mannucci, B.; Albini, A.; Fagnoni, M. "α,n-Didehydrotoluenes by Photoactivation of (Chlorobenzyl)trimethylsilanes: An Alternative to Enyne-Allenes Cyclization" Angew. Chem. Int. Ed. 2012, 51, 8577-8580. DOI: 10.1002/anie.201202794
- Ravelli, D.; Protti, S.; Fagnoni, M.; Albini, A. "From Phenyl Chlorides to α,n-Didehydrotoluenes via Phenyl Cations. A CPCM–CASMP2 Investigation" J. Org. Chem. 2013, 78, 3814-3820. DOI: 10.1021/jo400269s
Aromatic Sulfonates and Phosphates as PhotoAcid Generators (PAGs)
The study of novel photoresists is of paramount importance in several application-oriented fields, ranging from microelectronics to photonics, nanofabrications, sensors development. In whatever domain the realisation of micro/nanostructures on photoresist is required, a high spatial resolution combined with a high sensitivity is strongly pursued, together with the possibility of adding new photoresist features.
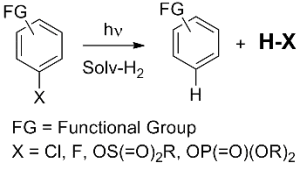
Since several years we have been involved in the study of possible photoinitiators. It was demonstrated that the irradiation of suitable aryl halides, sulfonates or phosphates (ArX) caused an efficient release (ca. 100%) of the corresponding strong acids (HX) such as HCl, sulfonic and phosphoric acids by heterolysis of the Ar-X bond. It is thus possible to tune the strength of the photogenerated acid by modifying the group X.
References
- Terpolilli, M.; Merli, D.; Protti, S.; Dichiarante, V.; Fagnoni, M.; Albini, A. "Cationic and Radical Intermediates in the Acid Photorelease from Aryl Sulfonates and Phosphates" Photoch. Photobio. Sci. 2011, 10, 123-127. DOI: 10.1039/C0PP00284D
- For more details on the other research lines in our lab, go to the COLLABORATIONS section